
For years, a heated debate has roiled the genetic genealogy community about small DNA segments. Judy Russell has recently written an excellent summary of the issue.

The precise definition of “small” varies, but the scientific consensus is that segments less than 6–8 cM are not trustworthy for genealogy purposes. Such segments are likely to be “false positives”, meaning they look like real segments of DNA inherited from a shared ancestor (called “identical by descent”, or IBD), but they’re not.
Scientists from 23andMe even published a peer-reviewed paper about false-positive segments way back in 2014. They compared child–parent trios (where a child and both parents had tested) to other people and tracked how frequently other people matched the child but not either parent. That is, of course, biologically impossible. Those segments are false positives.

The smaller the segment, the worse the problem is, as this graph shows. That’s why most leading DNA companies ignore segments less than 6 cM.
Even so, some genealogists continue to use small segments as “proof” of relationships. One DNA matching company, FamilyTreeDNA, even reported segments down to 1 cM, adding a sheen of credibility to the idea that tiny segments are somehow valuable for genealogy research.
Until now.
FamilyTreeDNA Updates Their Matching Program
Recently, FamilyTreeDNA updated their matching algorithm to exclude all segments less than 6 cM. They also published a white paper explaining why.The key evidence is summarized in their Table 2.

Focus on the left-most and right-most columns. At 1 cM, almost all of the “matching” segments–99.96%–are false positives. Put another way, only four out of every 10,000 such segments are identical by descent (IBD), and there’s no way to tell which four are legit from looking at them. The situation is only marginally better for 2-cM and even 3-cM segments. Even at 5 cM, nearly half of all segments are false. And again, there’s no indication of which half are real.
Why the False Segments?
At this point, you may be thinking “But I thought DNA matching was hard science. How can so many segments be wrong?” And that’s a great question! We should always be asking why.
The reason is that the technology we use for genetic genealogy is not perfect. It’s extremely good, but it has a fundamental weakness: it can’t tell which bits of autosomal DNA we inherited from our mothers and which we inherited from our fathers. This causes a mix-and-match problem (pun intended) that leads to false segments.

Consider the example to the left. Person A and Person B each have two copies of the chromosome, one paternal (blue) and one maternal (red). Neither sequence (haplotype) from Person A matches either sequence from Person B. Even so, this region gives a false positive match because at every point, at least one base in Person A matches at least one base in Person B. The error is caused by “haplotype switching”, where the match only appears to exist because the computer analysis is switching back and forth from one haplotype to the other.
Yes, newer sequencing technology that can get around the haplotype-switching problem, but it is both expensive and error prone. Our current methods may be imperfect, but they’re affordable, and there’s a simple workaround: ignore the segments that are statistically likely to be false.
Which is precisely what FamilyTreeDNA is now doing. As of August 2021, they only consider segments of 6 cM or more. This may cause substantial changes to your Family Finder match lists, but it’s a long-overdue change. It should also better align the DNA matches there with those in the other databases.
Worse Than It Looks
The false-positive problem is even worse than the data above suggest. That’s because FamlyTreeDNA’s Table 2 is based on simulations.

There were no mis-calls or no-calls or imputation errors in the data. There was no pedigree collapse. And the tree was known with absolute certainty, so the false positive segments were easy to spot. Even in that best-case scenario, segments below 5 cM were more likely to be false than true.
In the real world, there will be more false positives, because real data isn’t perfect. Some bases are called incorrectly. Some aren’t called at all. What’s more, companies that accept uploaded data files, like FamilyTreeDNA and MyHeritage, have to account for the fact that those files are not fully compatible with their own tests. They use a statistical trick called “imputation” to get around the incompatibility, but imputation can introduce its own errors. All of those errors can increase the false positive rate.
The Population Problem
Layer on to that one more issue: some segments really are IBD but don’t reflect a recent common ancestor. Instead, they are shared because they were widespread in the ancestral population. This often happens because the ancestral population was endogamous and the members were all related to one another. Ultimately, all ancestral populations were endogamous, so we all need to be concerned about small segments misleading us.
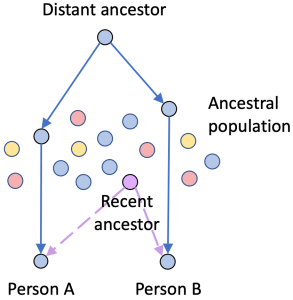 Consider this scenario: Person A and Person B share the “blue” DNA segment (solid path), and both also have “Purple” in their trees (dashed path). While it might seem reasonable to say the blue segment is proof that A and B are both descended from “Purple”, that would be wrong. The shared DNA came to both A and B through a much more distant ancestor via a population that had lots of blue descendants.
Consider this scenario: Person A and Person B share the “blue” DNA segment (solid path), and both also have “Purple” in their trees (dashed path). While it might seem reasonable to say the blue segment is proof that A and B are both descended from “Purple”, that would be wrong. The shared DNA came to both A and B through a much more distant ancestor via a population that had lots of blue descendants.
In this case, three statements are true:
-
- A and B might both be descended from Purple.
- A and B share a “blue” segment of DNA.
- The “blue” segment is not evidence that A and B are descended from Purple.
In other words, the small IBD segment in this case might be proof that A and B are related but it’s not proof of how they are related.
And that conclusion can only be drawn after you have confirmed that the segment is IBD in the first place.

Well, in general I agree however I’d like to make two points:
– The last example of a distant ancestor and the purple ancestor is completely ignoring that we can use the length of the matching segment to evaluate how far the common ancestor is. Itzik Pe’er has a good presentation on this with this “The genome of the Netherlands” paper.
– Some companies use very good algorithms to identify IBD segments and keep the false positive rate. Hint, FTDNA isn’t one of them, neither is MyHeritage IMO. The key lies in the effort one puts into identifying a segment that is phased (even without parent/child data). Ancestry DNA is clearly leading in this regard, 23andMe is second IMO
How would you use segment length when all of the segments are small?
Leah, thank you for this information and keep us updated.
Because I am 78 years old, in the past some of my 6 cMs segments have been fruitful in finding tree matches. And yes, I know a tree match is nothing more than that, until a TG is done. I will look at a 6 cMs match if she is a generation, or several, younger than I. Because, perhaps, if one of her parents had tested, I would have had a 12 cMs match with a parent (factoring in recombination).
One of my ancestral grandfathers had two wives. Those cousins who were descended from wife #1, shared adequate cMs amongst themselves ; but I, who descended from wife #2 showed a lower cMs with THEM because they were descending from grandfather AND grandmother and I was descending from just the grandfather in my comparison to them. (I did not explain that very well).
Small segments can absolutely help in finding tree matches. The problem is that they don’t prove that the tree match is correct. Even triangulation won’t prove that. Consider the last diagram in the post. There will be other people who descend from Purple who will triangulate on the blue segment, because the descendants of Purple are also likely from the same population. It’s so challenging!
Yes, frustratingly, some things in genealogy may never be proven.
I was able to solve a 135-year-old mystery involving the NPE birth, under a different name, in a different county, of a great-grandmother via DNA. It took years until an appropriate match showed up, who then matched my great-aunt as 2nd to 3rd cousin. Since then, nearly everyone in the two downstreams: my gg-grandmother’s illegitimate daughter and a “legitimate” son – all share genetic matches. There will never be documented proof; the adoption took place in San Francisco before the earthquake and fire and the adoption was never recorded in the county of the adoptive parents. This will forever remain a 99% proven story. All times and places, in retrospect, align. (The birth father has never – yet? – been identified.)
HOWEVER, while I and two of my siblings match our closest living cousin on that side with 50 cM or more, one of my brothers shares no DNA with him. In another case, one of my sibs shares only 10 cM with a cousin that the rest of us have robust matches with. It’s given me much thought: what if those sibs were me and I had to pursue all of my research without those critical clues the rest of us were able to provide?
I extrapolate that to my father and his deceased siblings. Dad has fewer matches with some distant cousins than his own nieces and nephews do. Clearly his own siblings inherited some streams of DNA that he did not.
So when I look at another otherwise interesting (per context) small match I don’t reject it out of hand. I check it out, weigh it, set it aside for future reference, and let it stew. And often am grateful for the clue it provides.
Great work on the 135-year-old mystery! I think these older genealogical problems will benefit from comparing multiple testers (like you and your siblings) as a whole, because the amounts shared—even if it’s zero—can be informative in the aggregate.
Thanks Leah. Excellent explanation and summary. This is now my new and much-improved response to someone who asks me to invest in discovering a common ancestor based solely on tiny cM matches.
You’re welcome, and thank you for the compliment!
The problem is not the small segments, but how they are used. I would never call any DNA match “proved” on the basis of DNA alone. They are merely clues.
Remember the so-called vanity books of the late 1800’s? They were notoriously full of errors and bad claims. But if one sifted through carefully, they could point to avenues of research. We need all the info we can get to solve genealogical problems.
I was distressed when Ancestry culled out the smaller segments, because as I looked through my own matches, I saw some 8th cousins I would never have known about without the clues of these small matches.
As for the 10-20% of bad matches, I think that is about the level of many online trees.
Often, the problem is the small segments. If a segment is a false-positive, there is no “right” way to use it. Even a common ancestor in someone else’s tree doesn’t mean the segment is real or that it was inherited from that ancestor.
https://research.rug.nl/en/publications/whole-genome-sequence-variation-population-structure-and-demograp
Please also search for his YouTube, it’s very informative
Can you please summarize his findings?
When the data bases grow more, it will be easier to ignore the smaller cMs.
Absolutely! We’re already seeing that. Almost all of the kits I see at AncestryDNA have 10,000 or more matches. Those are mainly of European and African descent.
The FTDNA chart seems to have at least one glitch; it shows 12 as 0.55% (26 segments) and 13 as 0.61% (27 segments), which is backwards; it appears their sample size wasn’t large enough.
Good catch! I’d guess that’s a typo.
Totally agree, but they are still clues, in need of proof. I am sure every etective in the world knows about false clues.
Anyone who uses a small segment match on its own to establish a relationship needs their head read. However for people like me trying to establish relationships between possible cousins from an elusive noble ancestor at the limits of DNA matching they are invaluable. True, they are difficult to work with, but not impossible. You just need lots of patience.
You should never have to work with tiny segments. Even a 15-cM segment can date back dozens of generations. With a large enough pool of descendants from your elusive noble ancestor, you should find pairwise matches that are well above the threshold for false positives. From there, proving the relationship for all descendants would require a sophisticated statistical analysis.
DNA customer testing for genealogy is a deal with the devil.
In exchange for keeping the cost down, the analysts chop our DNA into tiny pieces, analyse them and then put the jigsaw back together using computer matching.
The longer the pieces are, the better the matching process goes.
Part of the definition of “IBD” is that the segment in question definitely comes from one or other parent. If we don’t have that parent’s DNA handy, then any assignment is an estimation. A fairly good one if we have the other parent. Or if the segment is reasonably long, the tails on either end should have a long overlap with other chopped up segments that we know come from a particular parent.
If we had just started at one end and analysed one base at a time, the analysis would be almost 100% accurate but prohibitively expensive.
Often the best we can do is some form of phasing. If we are lucky enough to be able to use both parents, small segments should be much more reliable.
But you won’t find the test labs telling you that until they offer phasing from parents.
The technology to ‘start at one end and analyze one base at a time’ for individual strands of DNA has only recently become available. Even the human genome project was unphased.
I understand your reasoning and would agree to not being definitive proof alone. However, when you do show as having a common ancestor, this improves that probability. Small segments should be available thru all Testing sites with a statement of probability and false positives. Using Ancestry service, no the small segments should not populate when using the List of DNA Matches. However, small segments should be used when using Thru-Lines, Triangulation, etc that uses other data such as Family Trees, etc to increase it’s probability. And you should have the ability to review that match, confirm and link it to your tree. I have confirmed many distant cousins that were less than 5 cM shared segments. Thru-Lines used to find them faster than me until they changed it above 8 cM. Even my previous “confirmed” matches cannot be pulled up which is sad.
No, finding a common ancestor in your tree does not increase the probability that the small segment is real. In fact, one could argue that two people descended from the same ancestral population are more likely to have false positives because of how population genetics works. The only thing that can increase the probability for small segments is phasing for both partners in the match.
Antionette,
Actually, the scenario you describe is more likely to garner you false positives. ThruLines and tree data is not a suitable method to buttress these conclusions you’ve made.
Excellent answer!
A way I think the small segment issue can be framed is to ask whether a small segment match (say 6-8 cM) can have any genealogical value if it also (1.) has a shared match with someone who has the same couple in their tree (say a ThruLines match), and (2.) has more shared matches with a cluster of matches associated with this same couple.
We are not necessarily confined to consideration of the exact same small segment when there are other shared matches – at Ancestry these are at least 20 cM. Even an apparently invalid short segment match could indirectly detect valid matches on a somewhat different segment on the same chromosome or on a different chromosome. I introduced the basic concept of indirect detection of shared matches in my Sep. 3, 2020 post on Kitty Cooper’s Blog entitled “The Small Matches at Ancestry are Gone!” An empirical example supported the concept.
In the FTDNA paper, the “Population Problem” that you raise in your last section really appears to be addressed explicitly by the FTDNA white paper. And the second of their five steps (see pages 4-5 for the overview and page 8 for the Endogamy Classification) uses a support vector machine classifier to specifically address the issue that you raise. This is akin to the Ancestry TIMBER algorithm to weed out excess IBD regions.
What FTDNA has done really is a positive breakthrough. Within their process, the 50-50 threshold at 7 cM now allows IBD vs Non-IBD accuracy at the 50-50 level down to 5 cM.
Taken with the recent work of Tim Janzen on using autosomal DNA for matches in the 1700s and the work by Martin McDowell and others in the Ballycarry DNA Project identifying maiden names via DNA, it is possible WITHIN THE RIGHT LIMITS to find strong evidence from small shared regions in projects with large numbers of test takers.
The whole issue of the lone researcher looking only at other lone kits as opposed to a well-run project with dozens of kits also has to be kept in mind. This is a “don’t try this at home” area for lone researchers. But Tim Janzen and Martin McDowell and now FTDNA have shown that small regions are usable in the right circumstances. Low probability does not mean no probability. It is — and should be — very demanding to make a case for using small regions, but it is wrong to throw them out as unusable.
But FTDNA has thrown out the small segments with this new algorithm. They were the last holdout reporting segments down to 1 cM, and many people still insist such segments are valid as a result. It’s good to see that change. Now, from an education perspective, the challenge will be getting the lone researchers to understand that a shared segment, even an IBD one, isn’t necessarily proof that you and your match both descend from the most recent shared name in your trees.
I haven’t had a chance to evaluate FTDNA’s endogamy classification carefully. I agree that it’s a major move in the right direction.
Two other important aspects of the FTDNA results merit awareness.
1 – There is nothing in the white paper nor in their method about phasing. All of the results that they present are for unphased kits.
2 – The 50-50 threshold level for IBD vs Non-IBD at 7 cM in the general population that has been the wisdom for so long still remains. But FTDNA shows that within the context of their methods, 7 cM is not 50-50 but 80-20 IBD vs Non-IBD.
For me, there are two parts to genetic genealogy – the DNA segment part and the genealogy part. They go together. I triangulate segments down to about 10cM – it helps me group Matches. However, I will work on the genealogy part with anyone – large segment, small segment, no segment. After all, roughly half of my true 4C will not share any DNA with me. I agree that small segments, on their own, cannot be used as evidence of a relationship. However, finding cousins (and sharing research) does not require that we must have shared DNA.
It may be time for some updates to this “classic” review. While issues certainly do exist & remain with “small segments”, they are proving more and more helpful in confirming distant heritage. This is especially true with the new Ancestry pro-tool. While I understand and agree with the limitations and warnings on using these, it is absolutely conclusive, to me, that using segments all the way down to 6cM are very very fruitful when using elaborate chromosome painting and match clustering. Researching selected single segment matches are quite often far more productive than the 20+cM multiple segment matches that frequently get very obscure from multiple shared ancestors.
I disagree. Sub-7-cM segments still have the same problem with phasing and false-positives, even with chromosome painting and clustering. In addition, this recent post of mine highlights the skewed distribution of small segments (below 20 cM) in our trees. They’re much more likely to be very distant cousins than relatives in a genealogical timeframe. To use them for genealogy, you first have to rule out the alternate (and more likely) possibilities. https://thednageek.com/low-matches-lie/